

How is kilogram-scale production of Tirzepatide achieved? What segments are required?
Publish Time:
2025-08-19
This article is sourced from Eli Lilly Michael O. Frederick team, at June 17, 2021 Published in Org. Process Res. Dev
Abstract
Large-scale production of complex synthetic peptides faces many challenges, including manufacturing risks (such as product specification failures) and generally low yields and purity. To address these issues, we developed a hybrid solid-phase peptide synthesis / liquid-phase peptide synthesis ( SPPS/LPPS ) method for the synthesis of Tirzepatide. Through continuous production and real-time analytical monitoring, high-quality product manufacturing was ensured, while nanofiltration technology enabled purification of intermediates, avoiding complex precipitation steps. The implementation of this strategy was highly effective, resulting in a robust process with high yield and purity.
Introduction
Synthetic peptides are an important drug form, increasingly used in the treatment of various diseases. Synthetic peptides with molecular weights between 1000–5000 Da are much larger than typical small molecules (usually 200–600 Da ) but much smaller than large molecules such as monoclonal antibodies (usually greater than 50,000 Da ), belonging to a unique " medium molecular weight ” category. Synthetic peptides present both synthetic challenges and opportunities for the development and application of new technologies. This article summarizes the development process of a novel synthetic route for the phase III candidate drug Tirzepatide, including the use of a hybrid III coupling strategy to assemble the molecule, nanofiltration to purify intermediate solutions, and continuous production technology combined with real-time analytical data to ensure high-quality product manufacturing. SPPS/LPPS The implementation of this strategy was highly effective, resulting in a robust process with high yield and purity.
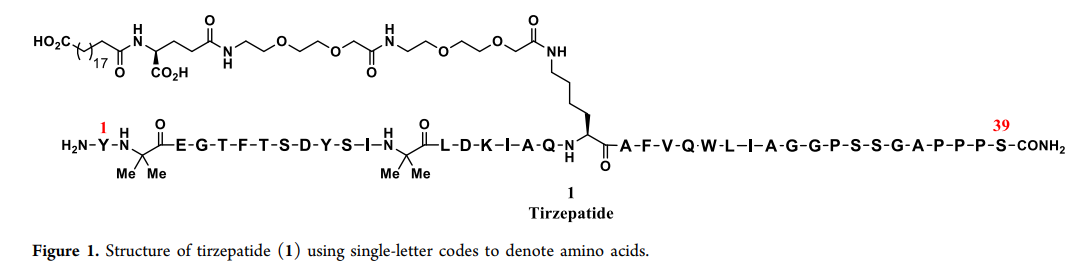
Tirzepatide Structure
Tirzepatide ( 1,figure1 ) is a novel GIP (gastric inhibitory polypeptide) and GLP-1 (glucagon-like peptide '1' ) dual receptor agonist used to treat diabetes, non-alcoholic steatohepatitis ( NASH ) and chronic weight management. This molecule is currently in III phase II clinical trials, with phase II studies showing significant potential in lowering HbA1c levels and reducing weight, promising to become a leading incretin therapy. Tirzepatide consists of a peptide chain of 39 amino acids and side chains at the 20 position residues, among which 37 are natural amino acids, and the 2 and the 13 positions are non-coded amino isobutyric acid residues. Given the potential of this molecule, a scalable and robust manufacturing process needs to be developed.
There are various methods for manufacturing synthetic peptides, table1 summarizing some synthetic methods of recently approved drugs. The choice of synthetic method is usually based on molecular features such as the presence of non-coded amino acids, non-peptide side chains, and the number of amino acids.
Recombinant technology commonly used for large molecules (such as monoclonal antibodies) can also be applied to smaller peptides. This method involves transferring exogenous DNA into Escherichia coli or mammalian (Chinese hamster ovary, CHO ) cell lines, where peptides are synthesized by the cells. Although development time is usually longer than typical synthetic methods, this approach has been successfully used for semi-synthetic peptides (such as semaglutide, degludec insulin, and liraglutide). Two drawbacks of this method are: ( 1 ) difficulty in introducing DNA non-coded amino acids; ( 2 ) difficulty in introducing non-peptide side chains.
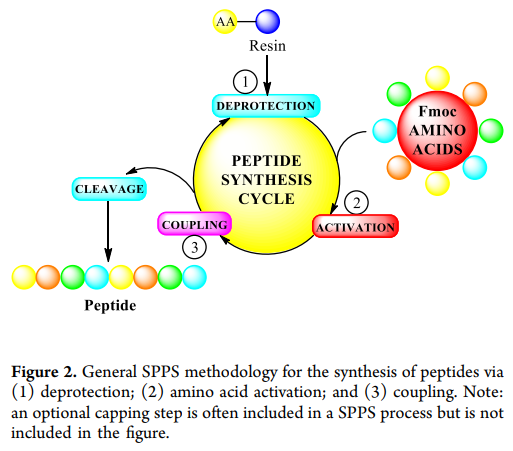
Another common method is solid-phase peptide synthesis ( SPPS ). This method was first proposed by Merrifield in 1963 year, synthesizing peptide chains by stepwise coupling of amino acids on resin. A typical SPPS cycle ( figure2 ) includes: ( 1 ) deprotection (usually removing Fmoc protecting groups with piperidine); ( 2 ) activation of the next amino acid; ( 3 ) coupling the activated amino acid to the growing peptide chain on the resin. Repeating this cycle synthesizes protected peptides, which are then cleaved from the resin and deprotected to obtain the target peptide (in some cases, soft cleavage " retains protected peptides or peptide fragments). Early ” use of SPPS 使用 N-α-Boc Protected amino acids as key components require large amounts each cycle HF Removal Boc group. To avoid using HF , recently 20 years researchers have shifted to using N-α-Fmoc protected amino acids, with piperidine for deprotection. SPPS It has been successfully used for various medium-sized molecules (such as liraglutide and exenatide), including the introduction of non-coding amino acids during synthesis. However, SPPS when synthesizing longer peptide chains (usually >30 amino acids), the yield and purity are lower, and due to the need for continuous execution of many unit operations, there is a higher manufacturing risk ( >30 amino acid peptide chain failure rate is estimated at 20% ).
Another common method is liquid-phase peptide synthesis ( LPPS ). This method is similar to SPPS , but the growing peptide chain is not attached to resin, and the peptide's C -terminus is a non-reactive amide or protected ester. This method is suitable for smaller peptide chains ( ≤10 amino acids, such as degarelix), but as the peptide chain grows, yield and purity rapidly decrease.
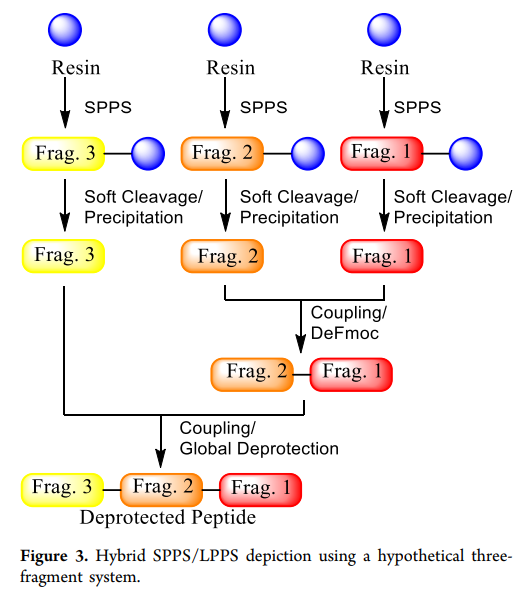
The last option is a hybrid SPPS/LPPS method (hereinafter referred to as " hybrid method ” ). This method synthesizes high-purity short peptide fragments as intermediates, avoiding manufacturing risks and purity issues in long-chain SPPS synthesis. Subsequently, these protected fragments are coupled in liquid phase ( SPPS shows a hypothetical three-fragment hybrid synthesis schematic). Although this method has been less used in the past (such as enfuvirtide), we consider it attractive. LPPS ,figure3 To implement the hybrid process, the primary task is to select fragments suitable for coupling by
. Key considerations for fragment selection include peptide chain length, total number of fragments, characteristics of the cleavage site amino acids, and side chains. The size and number of fragments are important because more small fragments, although purer, increase manufacturing burden and LPPS steps; fewer large fragments reduce manufacturing burden but increase purity and manufacturing risk. Considering these factors, we selected four fragments for the synthesis of telaprevir. For cleavage sites, a key factor is the racemization tendency of the amino acid at each fragment's LPPS terminus, because the α- C stereocenter of the amide backbone is more prone to racemization than the protected amino acids used in SPPS . After exploring various fragments, four fragments were finally selected ( Fmoc 2–5 ). These fragments are all easily separable solids with very high purity ( ,figure4 97.5–99.5% ). Preliminary studies using four fragments on a small scale ( ).
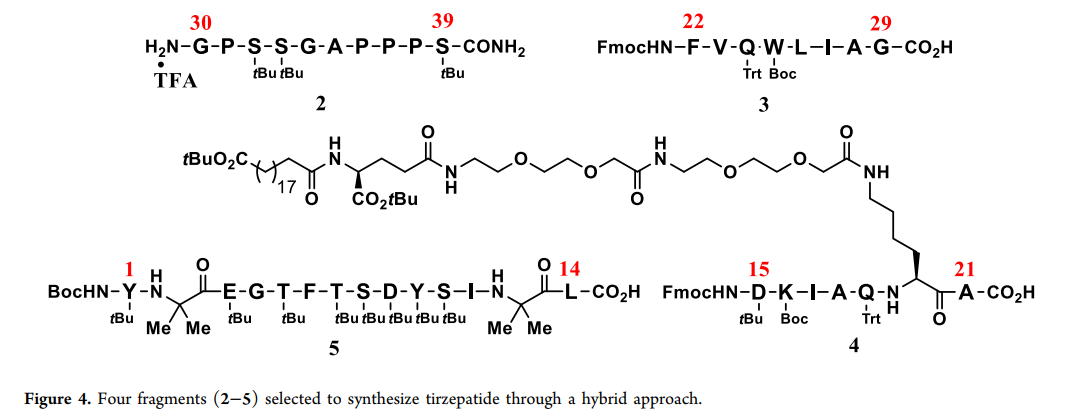
) verified the feasibility of the hybrid method. The study found that before scaling up to kilogram-level production, two new technologies can significantly improve peptide manufacturing. The first technology is using tangential flow filtration (especially nanofiltration) to purify intermediates, avoiding precipitation steps because long peptides tend to form gels and are difficult to separate. The second technology is continuous production combined with online high-performance liquid chromatography ( ). These fragments are all easily separable solids with very high purity ( HPLC ) analysis to ensure high-quality product manufacturing, reduce manufacturing risks, and decrease waste and costs. The production goal this time is to complete kilogram-level telaprevir crude synthesis in four batches. The first step of the process is coupling fragments
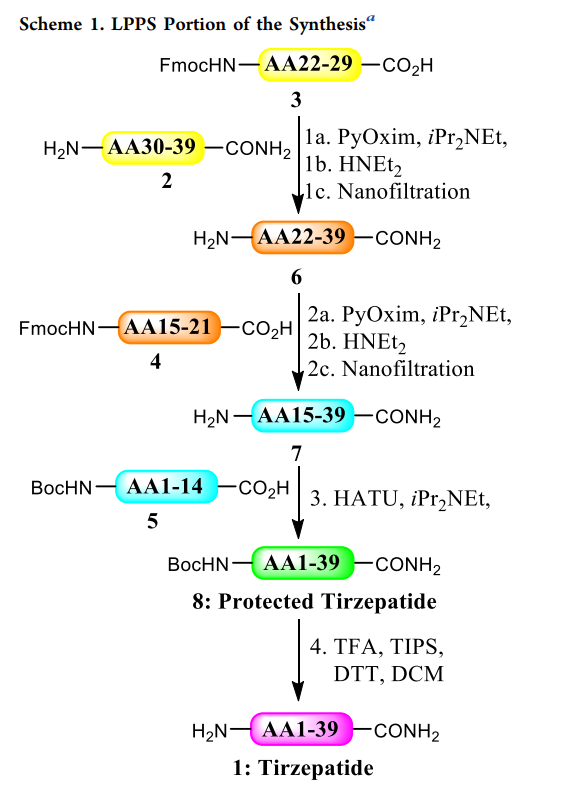
and 2 (representing amino acids 3 30–39 22–29 (representing amino acids ) respectively (all steps see LPPS ). The solutions of fragment scheme1 are dissolved in dimethyl sulfoxide ( 2 (representing amino acids 3 DMSO ) and acetonitrile ( ACN ), PyOxim is dissolved in , with pure diisopropylethylamine added, mixed through the flow system before entering the plug flow reactor ( ), PFR ) to ensure homogeneity ( equipment setup of step1 see figure5 ).
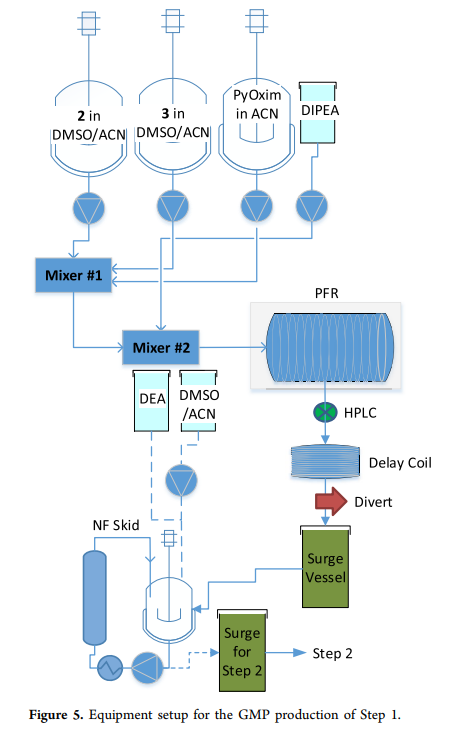
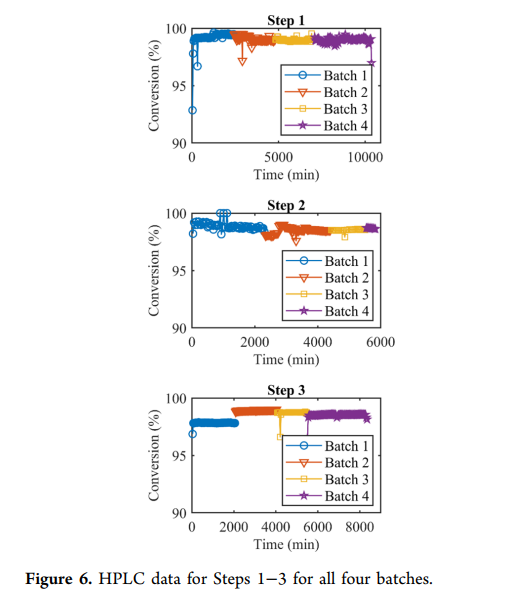
Coupling reactions in the flow system are tested during the process, allowing real-time monitoring of reaction quality and adjustment of stoichiometry. Materials meeting reaction specifications enter a buffer tank awaiting the next step; non-conforming materials are diverted (ultimately discarded), and by adjusting the flow rates of different materials, the materials collected in the buffer tank all meet quality requirements (note that no diversion occurred in the three coupling steps , all materials met process targets). step1 The solution of ) to ensure homogeneity ( in the plug flow reactor ( 3 ) has a residence time of 36 Minutes pass ) analysis to ensure high-quality product manufacturing, reduce manufacturing risks, and decrease waste and costs. Sample once. Remaining after sampling 45 Minutes reaction time, used to analyze the sample to ensure material quality, then the solution enters the buffer tank (production activity ) analysis to ensure high-quality product manufacturing, reduce manufacturing risks, and decrease waste and costs. Results see figure6 ). Another method is sampling at the end of the plug flow reactor ( ) to ensure homogeneity ( ) and installing a delay coil, whose length exceeds the time required for analysis. After the buffer tank is filled, the solution is deprotected with diethylamine ( DEA ) ( 20 equivalent, 1 hours) followed by nanofiltration to remove diethylamine, diethylamine - Dibenzofulvene ( DBF ) adducts and coupling-related impurities. The purified DMSO/ACN solution (containing amino acid 22–39 fragments 6 ) is stored in another buffer tank, waiting for step2。
Nanofiltration is a membrane-based technology that separates components in a solution based on molecular weight and hydrophobicity. After the crude reaction solution passes through the membrane, low molecular weight impurities (such as dibenzofulvene ( DBF ), diethylamine ( DEA ) and is dissolved in related by-products ( PyO )) are removed, while the target peptide is retained ( figure7 ). Similar technologies (such as tangential flow filtration and ultrafiltration) are commonly used for large molecule drugs (such as monoclonal antibodies), but ultrafiltration membranes are usually not suitable for organic solvent systems and short peptide fragments. Due to the larger molecular weight of peptides ( >2000 Da ), nanofiltration becomes feasible. Many nanofiltration / ultrafiltration membranes are incompatible with organic solvents, but some ceramic-based membranes can tolerate ) and acetonitrile ( 、 ), 、 DMF (representing amino acids DEA . The choice of pore size needs to balance low flux (pore size too small) and high peptide loss (pore size too large).

step1 Among them, 200 Da membranes with pore size (manufacturer Inopor calls its pore size <0.9 nm ) can achieve lower peptide loss. The product is first concentrated to reduce volume and circulation time, and a solvent volume of 8 times through the nanofilter is sufficient to remove low molecular weight impurities (such as diethylamine, dibenzofulvene and is dissolved in related impurities). ) and acetonitrile ( (representing amino acids ), The solvent rinse liquid is circulated and added to the final product to recover peptides on the membrane, and the final solution is diluted to the target concentration for the next step. The final purified fragment 6 ( AA22–39 ) solution, with a total yield of the three-step operation of 75–80% 。
step2 conditions are similar to step1 Using a plug flow reactor ( ) to ensure homogeneity ( ) to couple fragment 6 (containing amino acid 22–39 ) and fragment 4 (containing amino acid 15–21 ) in DMSO:ACN solution with iPr2NEt (representing amino acids is dissolved in at 20°C coupling, with a residence time of 3 hours. Through online ) analysis to ensure high-quality product manufacturing, reduce manufacturing risks, and decrease waste and costs. monitoring, high conversion rates in four batches of production are ensured ( figure 6 ). After coupling, the protecting group is removed with diethylamine, and the crude solution is purified by nanofiltration. Using Fmoc ceramic membranes with pore size ( 450 Da 0.9 nm pore size, since fragment molecular weight is higher than fragment 7 , a larger pore size can be used) to remove low molecular weight impurities (diethylamine, dibenzofulvene and 6 related by-products), while replacing the solvent with dimethylformamide ( is dissolved in ), obtaining purified fragment DMF 15–39 7 (containing amino acid 68–75% ) solution, with a total yield of the three-step operation of conditions are ineffective in 。
step1 (representing amino acids 2 due to the high racemization level of leucine residues in fragment step3 1–14 5 (containing amino acid ). After extensive optimization, finally using HATU with and a small amount of (for feed, as this reagent is unstable in iPr2NEt at DMF at ), 0°C (for 进料,因该试剂在 DMF 中不稳定)中于0°C Reaction below. The reaction mixture passes through a plunger flow reactor ( ) to ensure homogeneity ( ), with a residence time of 3 hours, aiming for complete conversion and D- leucine isomer content below 0.5% . Then purified by precipitation, because step3 purification cannot use nanofiltration (solvent replaced by TFA , mainly used for step4 ). Fortunately, using NaCl (representing amino acids NaHCO3 aqueous solution can successfully precipitate, after separation and drying obtaining the protected telaprevir ( 8 PyOxim step3 yield is 95–100% 。
The final step is to remove the 8 acid-labile protecting groups on the fragment 19 . Standard SPPS cleavage and deprotection conditions are used, employing TFA 、 TIPS and dithiothreitol (used to react with the cleaved protecting groups) as well as DCM and water (to increase solubility) at 21°C reaction 3 hours, completely removing all protecting groups. By adding methyl tert-butyl ether ( MTBE ) to precipitate the crude telaprevir ( 1 ). To avoid spherical agglomeration, the feeding rate must be strictly controlled and carried out in two stages: initial slow feeding ( 90 minutes adding 40% ) to minimize heat release, followed by rapid feeding ( 45 minutes adding the remaining 60% ) to avoid agglomeration observed during development. After separation and drying, the crude telaprevir is obtained 1,step4 yield is 75–85% 。
The mixing strategy aims to reduce manufacturing risks of telaprevir and achieve a process with high yield and high crude purity. All targets were met, and the process performed robustly in production (conversion data for three coupling steps see figure6 ), the total yield of the synthesized peptide is high (crude yield 46% ), and crude purity exceeds 70% . Unlike small molecule drugs purified by crystallization API , peptides require chromatographic purification to remove related peptide impurities. Higher purity of the crude API can simplify purification steps and improve overall yield.
In addition, mixed impurities are mainly caused by excess fragment stoichiometry, which can be easily removed by chromatography, further simplifying the purification process. The modular mixing method also facilitates optimization, and significant improvements have been achieved since this production activity ( LPPS yield >55% , crude purity >80% ).
Conclusion
This article describes the development and implementation of a mixing SPPS/LPPS strategy for kilogram-scale production of telaprevir. Through new technologies such as continuous production, real-time analytical monitoring, and nanofiltration intermediate purification, high-quality product manufacturing was achieved. This success provides potential application value for future peptide drug production.
Tongjun Pharmaceutical synthesizes and produces multiple fragments of telaprevir, including CAS:2682040-93-1, CAS:3034670-52-2, CAS:2656383-23-0, CAS:2461524-68-3, CAS:2656383-24-1, CAS:2656383-25-2, providing high-quality raw materials at competitive prices. Welcome to contact us!




